Revisión de la fisiopatología, diagnóstico y seguimiento: últimas actualizaciones de la Enfermedad Celíaca
Revisión de la fisiopatología, diagnóstico y seguimiento de las últimas actualizaciones de la Enfermedad Celíaca.
Un documento elaborado por la Comisión de Inmunología de CLILAB.
La celiaquía o enfermedad celíaca (EC) es una enfermedad inflamatoria crónica de origen autoinmune que afecta a la mucosa del intestino delgado y se desencadena por el consumo de gluten en personas genéticamente predispuestas, causando atrofia de las vellosidades intestinales, incremento del número de linfocitos intraepiteliales e hiperplasia de las criptas del intestino delgado.
La EC es más frecuente en mujeres que en hombres (relación 2:1) y presenta una prevalencia mundial en torno al 1 %.
Aunque se han descrito manifestaciones extraintestinales, las manifestaciones clásicas afectan al intestino, hasta el punto de que actualmente se considera la principal causa de malabsorción; por ello, es importante un diagnóstico precoz.
2. Fisiopatología de la enfermedad celíaca
El gluten está formado por un conjunto de proteínas presentes en las semillas de muchos cereales como el trigo, el centeno, la cebada, algunas variedades de avena o en cualquiera de sus variedades o híbridos (por ejemplo, espelta, escanda o kamut). No está presente en el maíz ni en el arroz.
Las principales proteínas del gluten son:
- Las prolaminas, que según su origen se clasifican en gliadinas (trigo), secalinas (centeno), hordeínas (cebada) y aveninas (avena), ricas en aminoácidos como la glutamina y la prolina.
- Las gluteninas, denominadas gluteninas cuando proceden del trigo.
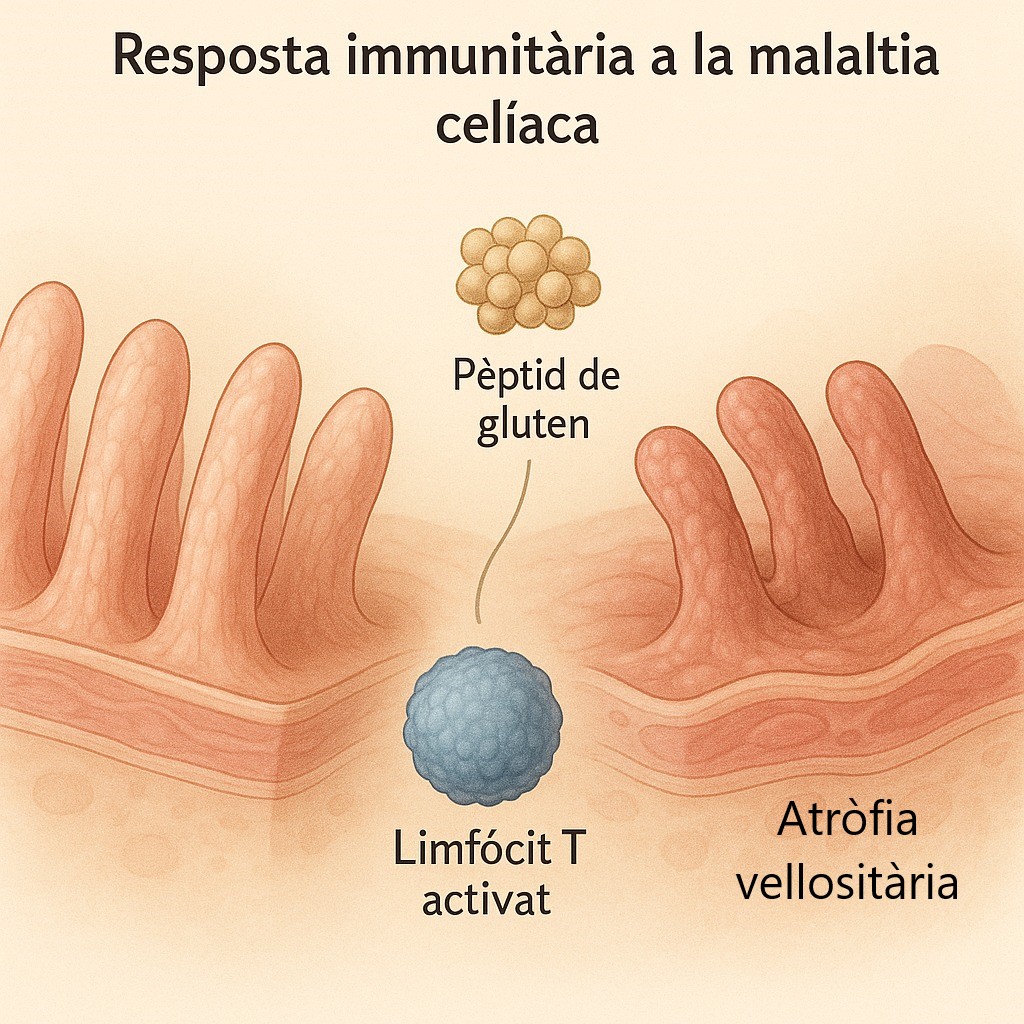
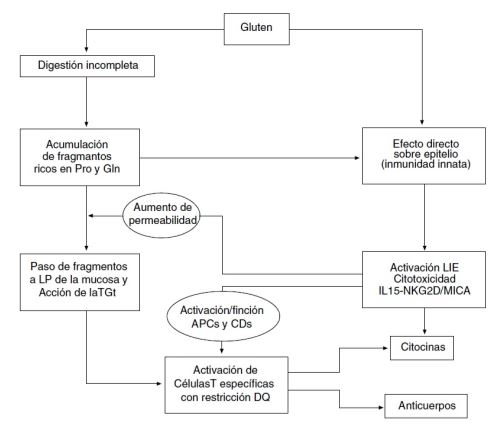
Los residuos de prolina de las gliadinas impiden su degradación proteolítica durante su paso por el páncreas, de modo que alcanzan intactas la lámina propia del intestino, donde son desaminadas por la enzima transglutaminasa tisular. Este proceso de desaminación modifica las gliadinas generando péptidos deaminados de gliadina con carga negativa, lo que facilita su unión a las moléculas HLA-DQ2 o HLA-DQ8 expresadas en las células presentadoras de antígeno de la lámina propia, responsables de presentar estos péptidos a los linfocitos T CD4.
Además, la interacción entre las gliadinas y la transglutaminasa tisular durante la desaminación produce una unión covalente entre ambas, generando complejos de mayor tamaño que también se unen a las moléculas HLA-DQ2 o HLA-DQ8. Por tanto, dichas moléculas pueden presentar a los linfocitos T CD4 tanto los péptidos deaminados de gliadina como los complejos gliadina‑transglutaminasa y, en algunos casos, neoantígenos derivados de su formación. La presentación de cualquiera de estos antígenos activa a los linfocitos T CD4, generando un ambiente proinflamatorio con producción de citocinas como IFN‑γ e IL‑21, que inducen la respuesta celular Th1 y Th2, así como la respuesta humoral al activar linfocitos B, productores de autoanticuerpos dirigidos contra los péptidos presentados por HLA‑DQ2 y HLA‑DQ8.
Existen péptidos de gliadina que no son reconocidos por linfocitos T pero interactúan directamente con el epitelio intestinal activando mecanismos de inmunidad innata, como la producción de IL‑15, que induce la apoptosis de los enterocitos, altera la función de barrera epitelial y aumenta la permeabilidad mediante la activación y proliferación de linfocitos intraepiteliales (LIE) CD8+ que expresan receptores de células NK, expresados por los enterocitos en situaciones de estrés.
La figura 1 resume los principales procesos patogénicos implicados en el desarrollo de la EC.
En conjunto, se genera un entorno altamente inflamatorio en la luz intestinal que provoca una alteración de la permeabilidad y de la absorción de nutrientes, dando lugar a las manifestaciones intestinales clásicas.
3. Manifestaciones clínicas
En función de la edad de presentación y de la clínica se distinguen:
- Formas clásicas o típicas: generalmente aparecen entre los 6 y los 18 meses de edad y se caracterizan por sintomatología intestinal derivada de la alteración de la permeabilidad y la malabsorción de nutrientes, causando pérdida de peso, diarrea (con o sin esteatorrea) y retraso del crecimiento.
- Formas no clásicas o atípicas: suelen aparecer en la edad adulta y los síntomas son muy variables, desde afectación intestinal (dolor abdominal, estreñimiento, distensión, síndrome del intestino irritable) hasta afectación extraintestinal (manifestaciones neurológicas, fatiga, irritabilidad, anemia ferropénica, dermatitis herpetiforme, osteoporosis, vómitos, infertilidad, abortos de repetición, etc.).
- Formas silentes: hay alteración de la histología intestinal y se detectan autoanticuerpos.
- Formas potenciales: no se detectan autoanticuerpos ni hay alteración de la histología intestinal; solo se demuestra una predisposición genética.
Entre el 1 y el 1,5 % de los pacientes diagnosticados pueden desarrollar formas refractarias, caracterizadas por la persistencia de síntomas (malabsorción y pérdida de peso) y de la atrofia vellosa tras un año de dieta estricta sin gluten. Las formas refractarias se dividen en dos subtipos según la presencia de subpoblaciones aberrantes de LIE: subtipo 1, sin subpoblaciones aberrantes; subtipo 2, con subpoblaciones aberrantes que representan un 20 % del total de LIE y se asocia a complicaciones graves como el linfoma T asociado a enteropatía.
4. Marcadores utilizados para el diagnóstico de EC
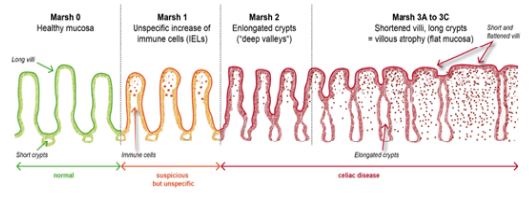
La biopsia intestinal ha sido el método de confirmación diagnóstica desde la década de 1950. Tradicionalmente se requerían tres biopsias: una con dieta con gluten; una segunda tras un periodo de dieta sin gluten; y una tercera después de una prueba de provocación con gluten para demostrar el empeoramiento con su ingesta. En 1990 la ESPGAN (European Society of Paediatric Gastroenterology and Nutrition) publicó los primeros criterios diagnósticos basados en la evaluación histológica de estas tres biopsias. Estudios posteriores demostraron que una única biopsia en el momento del diagnóstico era suficiente en el 95 % de los casos. Para la evaluación histológica se recomienda obtener varios fragmentos (1–2 del bulbo y 4 del duodeno distal) y utilizar la clasificación de MARSH, que permite clasificar el estadio de la enfermedad según el grado de atrofia vellosa e infiltración linfocitaria (Imagen 1).
Posteriormente aparecieron marcadores serológicos (anticuerpos) con buenos valores de sensibilidad y especificidad que han llevado a plantear, en las guías más recientes, un algoritmo diagnóstico sin biopsia. Estos anticuerpos son de isotipo IgA, salvo en pacientes con déficit de IgA (la inmunodeficiencia primaria más frecuente), en los que se determinan IgG. Los anticuerpos descritos se dirigen contra la isoforma tipo 2 de la enzima transglutaminasa tisular presente en la pared de los músculos lisos y en la mucosa del intestino delgado; y también contra los péptidos deaminados de gliadina, con mayor sensibilidad en niños menores de 12 años. Además, en el algoritmo sin biopsia propuesto en las últimas guías (véase el apartado 5) se incluyen los anticuerpos antiendomisio. El endomisio es un tejido conectivo que forma una vaina que recubre los músculos y en el que también está presente la transglutaminasa tisular.
En la formación de estos anticuerpos interviene la predisposición genética relacionada con genotipos concretos de los alelos HLA (ya mencionados en el apartado 2). Los alelos HLA son moléculas expresadas en la superficie de las células nucleadas que participan en la respuesta inmunitaria. Su función es presentar antígenos exógenos (o autoantígenos en enfermedades autoinmunes) a los linfocitos T, induciendo la respuesta celular y la humoral con formación de anticuerpos. Los genotipos de susceptibilidad descritos para desarrollar EC son: HLA‑DQ2.5, HLA‑DQ2.2, HLA‑DQ7.5 y HLA‑DQ8. Aun así, su presencia no es diagnóstica, pues están presentes en el 35–40 % de la población general y solo un 1 % de estas personas desarrolla la enfermedad.
Cuando ni la histología (MARSH 0–1) ni los anticuerpos son concluyentes, ha cobrado importancia el estudio del inmunofenotipo de los linfocitos intraepiteliales (LIE) en la biopsia mediante citometría de flujo. Este estudio ofrece una sensibilidad aproximada del 92 % y una especificidad del 93 % para el diagnóstico de EC. Se ha descrito en pacientes con EC un incremento del número absoluto de LIE, un aumento del porcentaje de la subpoblación CD3+TCRγδ+ y una disminución de la subpoblación CD3–CD103+.
5. Revisión de las guías diagnósticas de EC
Existen varias guías para el diagnóstico y seguimiento de la enfermedad celíaca. La última actualización del Colegio Americano de Gastroenterología se publicó en 2023. Tanto esta actualización como la guía de la sociedad europea de estudio de la EC y la de ESPGHAN (European Society for Paediatric Gastroenterology, Hepatology and Nutrition) no contemplan cribado universal; recomiendan la búsqueda activa en pacientes con síntomas o en familiares de primer grado de pacientes diagnosticados. Las tres coinciden en que el estudio debe realizarse cuando el paciente mantiene una dieta con gluten.
Para el diagnóstico, las tres guías coinciden en iniciar con serología: anticuerpos antitransglutaminasa IgA junto con IgA total para detectar déficit (presente en el 2–3 % de los pacientes con EC). En caso de déficit, se realizará estudio serológico de isotipo IgG. En adultos con antitransglutaminasa IgA positiva, las tres guías recomiendan confirmar el diagnóstico probable mediante endoscopia con toma de múltiples biopsias duodenales (varios fragmentos del bulbo y del duodeno distal), como se indicó en el apartado 4.
ESPGHAN define un protocolo diagnóstico sin necesidad de endoscopia en niños; esta opción también la contempla la última actualización del Colegio Americano de Gastroenterología para adultos con condiciones fisiológicas que impidan la endoscopia o que no la acepten voluntariamente. En estos casos, si presentan anticuerpos positivos con niveles superiores a 10 veces el límite superior de la normalidad, se confirmará con una segunda muestra de sangre y la determinación de anticuerpos antiendomisio. La confirmación de niveles de antitransglutaminasa y la positividad de antiendomisio IgA en la segunda muestra permite clasificar a los adultos como diagnóstico probable de EC y, en niños, establecer el diagnóstico de EC. Respecto al estudio genético (HLA‑DQ2/DQ8), las tres guías coinciden en su elevado valor predictivo negativo, por lo que recomiendan solicitarlo en pacientes que iniciaron dieta sin gluten antes del estudio serológico, en casos con serología negativa pero histología sugestiva de EC, o en pacientes con clínica pero serología e histología negativas, en los que un resultado negativo permite prácticamente excluir el diagnóstico.
6. Seguimiento de la EC
El tratamiento de la EC es la dieta sin gluten (DSG). Es fundamental educar al paciente sobre la importancia de la adherencia para evitar déficits nutricionales, asegurar un correcto crecimiento y desarrollo en niños y lograr la restauración completa de la mucosa intestinal. ESPGHAN (niños y adolescentes) y el Colegio Americano de Gastroenterología (adultos) coinciden en el siguiente protocolo de seguimiento tras el diagnóstico:
- Evaluación clínica con presencia/ausencia de síntomas gastrointestinales o extraintestinales.
- Parámetros antropométricos y de crecimiento en población pediátrica.
- Anticuerpos antitransglutaminasa IgA (o IgG en caso de déficit) como marcador de curación mucosa y seguimiento de la DSG.
- Determinaciones bioquímicas para detectar déficits y enfermedades asociadas: hemoglobina, hierro, vitamina B12, vitamina D, metabolismo hepático (ALT), TSH, T4, anti‑TPO, anti‑TSH y antitiroglobulina.
En este contexto ha cobrado fuerza la determinación de péptidos inmunogénicos del gluten (GIP). Aunque ESPGHAN refiere falta de evidencia para recomendaciones firmes, la Sociedad Española de Enfermedad Celíaca (SEEC) publicó el pasado año un protocolo (“Evaluación de la adherencia a la dieta sin gluten en pacientes adolescentes y adultos con enfermedad celíaca: estrategia de manejo de los péptidos inmunogénicos del gluten”) sobre su utilidad en adolescentes (≥14 años) y adultos diagnosticados, para valorar adherencia a la DSG. Los GIP son fragmentos de gluten resistentes a la digestión gastrointestinal y principales responsables de la respuesta inmunitaria en celíacos. La detección de GIP en heces u orina indica consumo de gluten de forma no invasiva. Existen estudios que demuestran correlación entre la atrofia vellosa en la biopsia y la detección de GIP en heces u orina. La SEEC propone determinación semestral de GIP en pacientes bien diagnosticados. Una o más determinaciones positivas durante el seguimiento permite predecir la persistencia de lesiones histológicas y, en pacientes con síntomas persistentes, discriminar entre mala adherencia y coexistencia de otras entidades clínicas. Para GIP fecales se recomiendan dos muestras/semana, 2–3 días antes de la revisión; en orina, tres muestras/semana, recogidas con la primera orina del día.
7. Puntos clave
- Los anticuerpos dirigidos contra la transglutaminasa son los principales relacionados con la EC y se utilizan sobre todo en el inicio del diagnóstico y el seguimiento de la DSG. En CLILAB utilizamos la técnica de quimioluminiscencia (CLIA), de alta sensibilidad y especificidad.
- Para una correcta interpretación de los antitransglutaminasa es importante descartar previamente un déficit de IgA y mantener el gluten en la dieta hasta el diagnóstico definitivo.
- En CLILAB estamos alineados con las últimas guías de ESPGHAN y, por ello, disponemos de la determinación de anticuerpos antiendomisio, que se solicitan desde el laboratorio siguiendo el algoritmo propuesto.
- Recientemente, en CLILAB hemos incorporado el inmunofenotipo de LIE en biopsia duodenal mediante citometría de flujo, que permite confirmar o descartar el diagnóstico en casos dudosos.
- El último protocolo de la Sociedad Española de Enfermedad Celíaca (SEEC) otorga relevancia al control de transgresiones dietéticas difíciles mediante la determinación de GIP en heces y orina. En CLILAB conocemos su utilidad y la hemos incorporado recientemente al catálogo.
Bibliografía
- Am J Gastroenterol 2023;118:59–76: American College of gastroeneterology Guidelines Update: Diagnosis and Management of Celiac Disease
- JPGN • Volume 75, Number 3, September 2022: ESPGHAN Position Paper on Management and Follow up of Children and Adolescents with celiac disease
- Sociedad Española de Enfermedad Celíaca: Evaluación de la adherencia a la dieta sin gluten en pacientes adolescents y adultos con enfermedad celíaca: estrategia de manejo de los péptidos inmunogénicos del gluten
- Novedades en el diagnóstico de la enfermedad celíaca. Pérez Torrella, D., Paniagua Arribas, E.; For Cont Lab Clin 2024;7;1-29
- Foods 2025, 14, 959: Celiac Disease—Narrative Review on Progress in Celiac Disease