Talasemias: importancia del diagnóstico diferencial

El diagnóstico de las talasemias es complejo debido a la diversidad genética detrás de sus síntomas. Diferenciar las talasemias de otras causas de microcitosis y anemia requiere una evaluación combinada de características hematológicas, bioquímicas y genéticas.
La hemoglobina (Hb) de los glóbulos rojos, o hematíes, puede estar afectada en diversas condiciones lo que repercute en su capacidad de transportar oxígeno. Estas enfermedades se conocen como hemoglobinopatías.
Estas pueden clasificarse en dos tipos principales:
- Hemoglobinopatías cuantitativas: producción reducida de alguna de las cadenas de globina. Esto provoca acumulación de las cadenas sobrantes y puede llevar a eritropoyesis ineficaz y hemólisis.
- Hemoglobinopatías cualitativas o estructurales: se forman variantes de hemoglobina con características fisicoquímicas alteradas. Un ejemplo es la hemoglobina S, responsable de la drepanocitosis.
Cada año nacen en todo el mundo aproximadamente 90.000 niños con síndromes talasémicos y son especialmente prevalentes en regiones del Mediterráneo, África y el sudeste asiático.
Las talasemias se caracterizan por la producción insuficiente o defectuosa de hemoglobina. Esto provoca una disminución del tamaño de los hematies (microcitosis) que puede causar anemia, eritropoyesis ineficaz y hemólisis de diversos grados.
El diagnóstico diferencial de talasemias supone un reto, ya que la microcitosis tiene múltiples causas, incluyendo la deficiencia de hierro.
Clasificación de las talasemias
La afectación o ausencia de los genes de una de las cadenas de globina, α o β, provoca un exceso de la cadena complementaria en la sangre. Según la cadena adectada pueden clasificarse en talasemias alfa (α) y beta (β). Además, según la severidad de los síntomas se clasifican en:
- Menor: generalmente asintomática y diagnosticada incidentalmente. En estos casos es importante el asesoramiento genético, ya que existe el riesgo de tener hijos con formas graves de la enfermedad si ambos progenitores son portadores.
- Intermedia: puede requerir transfusiones ocasionales y es más pronunciada en las talasemias beta. Es necesario realizar un seguimiento adecuado, ya que pueden desarrollar complicaciones relacionadas con la sobrecarga de hierro. Estas complicaciones pueden dañar órganos vitales, como el corazón y el hígado.
- Mayor: grave, con un pronóstico desfavorable, complicaciones severas y requiere de transfusión de sangre regulares. En el caso de las talasemias alfa o síndrome de Bart suele ser incompatible con la vida. En el caso de las talasemias beta se conoce como anemia de Cooley.
Diagnóstico de talasemias y microcitosis no ferropénica
Uno de los retos principales en el diagnóstico de las talasemias es la distinción entre microcitosis ferropénica y no ferropénica. La deficiencia de hierro es la causa más común de microcitosis, pero la talasemia es también una causa que puede presentar rasgos similares.
La talasemia suele estar acompañada por un número de hematíes alto mientras que en la anemia por deficiencia de hierro, el conteo de hematíes tiende a estar reducido.

Figura 1. Diferencias entre talasemias y anemia ferropénica
Los niveles bajos de hierro en el laboratorio no siempre indican deficiencia de hierro ni anemia. Aunque se suelen utilizar otros parámetros para evaluar el metabolismo del hierro estos pueden estar alterados por inflamación u otras condiciones.
La microcitosis no es exclusiva de las talasemias y puede estar presente con o sin anemia Otras condiciones, u hemoglobinopatías, también pueden presentar microcitosis. La clave para el diagnóstico es descartar otras causas y confirmar la presencia de la talasemia mediante análisis específicos de hemoglobina y estudios genéticos.
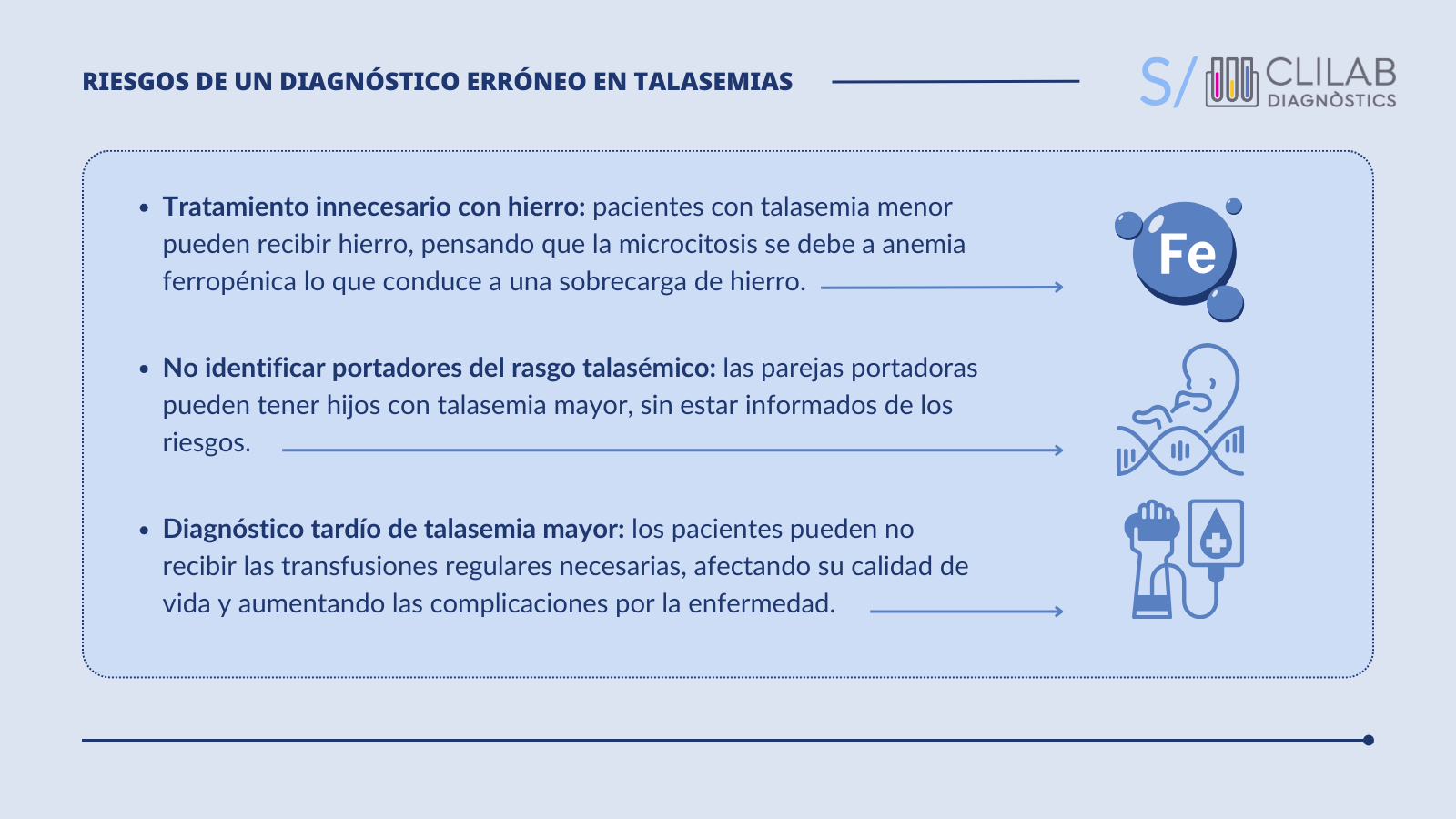
Figura 2. Riesgos de un diagnóstico erróneo en talasemias
Pruebas diagnósticas en el laboratorio clínico
Se combinan pruebas hematológicas, bioquímicas y genéticas. Algunas de las pruebas más relevantes son:
1. Hematología básica
Se empieza con un conteo sanguíneo completo (CBC) para evaluar la morfología de los hematíes. La microcitosis y la hipocromía son signos comunes. Sin embargo, también se observan en la anemia por deficiencia de hierro, lo que requiere pruebas adicionales.
La amplitud de distribución eritrocitaria (ADE), suele estar aumentada en la anemia por deficiencia de hierro, mientras que en las talasemias tiende a ser estrecho o normal.
Los pacientes enfermos o portadores de las alfa talasemias muestran alteraciones hematológicas desde recién nacidos mientras que las beta talasemias suelen aparecer a partir de los 6 meses.
Por lo tanto, si los antecedentes del paciente presentan un hemograma normal generalmente se puede descartar la talasemia como causa de la microcitosis.
2. Electroforesis de hemoglobina
Esta técnica permite identificar y cuantificar las diferentes fracciones de hemoglobina en la sangre del paciente. En la beta talasemia, los niveles de hemoglobina A2 (HbA2) están aumentados, mientras que la hemoglobina fetal (HbF) puede estar presente en cantidades variables. También se detectan hemoglobinas anormales como Hb Bart .
No obstante, es importante destacar que la deficiencia de hierro puede reducir falsamente los niveles de HbA2. Esto puede complicar el diagnóstico de la beta talasemia. Por esta razón, es importante tratar el estado de ferropenia antes de interpretar los resultados de hemoglobina.
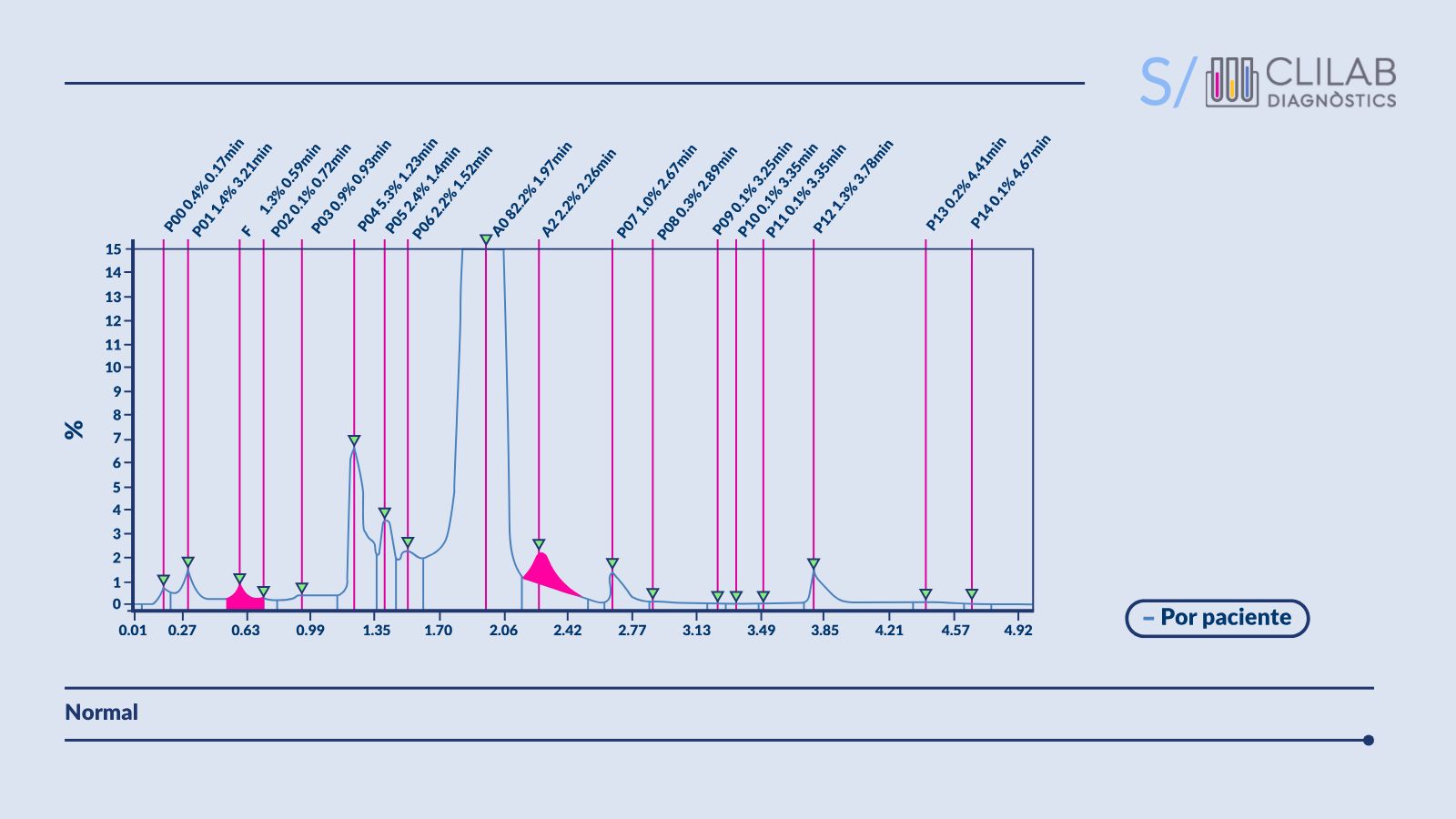
Figura 3. Cromatogramas HPLC de un paciente sin talasemia.
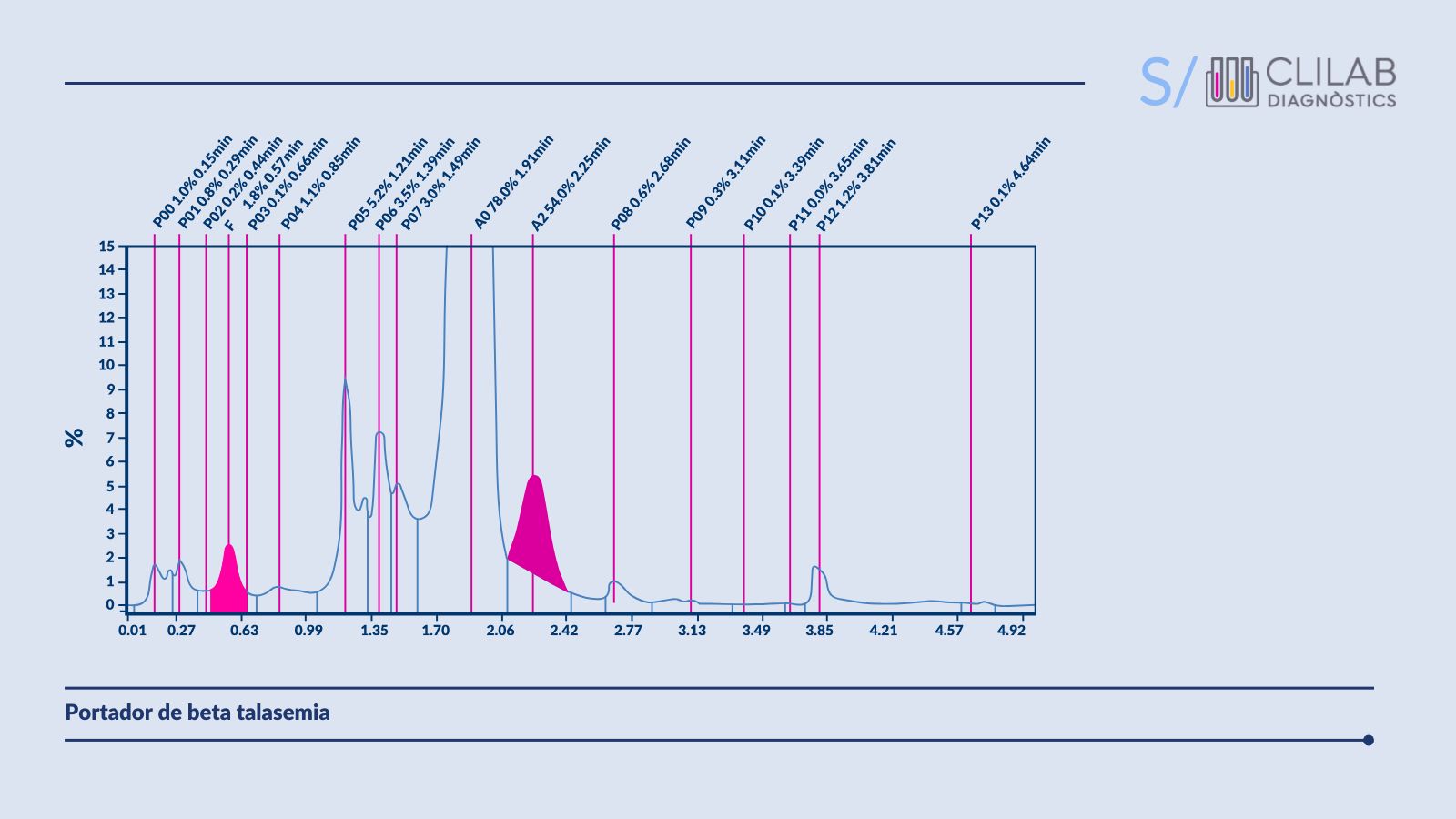
Figura 4. Cromatogramas HPLC de un paciente con talasemia.

Figura 5. Elementos esenciales para la evaluación de microcitosis en el laboratorio clínico
3. Cromatografía líquida de alta resolución (HPLC)
Sirve para detectar variantes de hemoglobina y anomalías en los niveles de HbA, HbA2 y HbF. Ofrece resultados rápidos y precisos, lo que facilita la identificación temprana de las talasemias y permite un seguimiento eficaz.
4. Análisis de ADN
Permite identificar las mutaciones específicas en los genes responsables de la producción de cadenas de globina y es el método más definitivo para el diagnóstico de talasemia, especialmente la alfa en portadores asintomáticos. Estos pacientes pueden presentar resultados normales en muchas pruebas hematológicas convencionales, lo que requiere de pruebas genéticas para confirmar el diagnóstico.
Las técnicas de diagnóstico genético como la reacción en cadena de la polimerasa (PCR) están cada vez más presentes en los laboratorios clínicos.

Figura 6. Genes estudiados en el análisis genético de talasemias
Conclusión
Los pacientes portadores heterocigotos o con formas leves, pueden ser diagnosticados erróneamente como anémicos por deficiencia de hierro. Ello conduce a un tratamiento con suplementos de hierro ineficaz y potencialmente perjudicial, ya que el exceso de hierro puede acumularse en el cuerpo, causando toxicidad.
Por ello, el diagnóstico diferencial de las talasemias, a través de pruebas hematológicas y genéticas, es esencial para identificar correctamente a los pacientes y portadores y orientar el manejo adecuado del paciente.
En CLILAB estamos comprometidos con la precisión diagnóstica con el objetivo de mejorar la calidad de vida de los pacientes con talasemia y reducir las complicaciones a largo plazo asociadas a esta enfermedad hereditaria.
Artículos relacionados:
- Trombofilia, ¿qué es y cómo se diagnostica?
- Diagnóstico del laboratorio de la anemia por deficiencia de hierro: herramientas y desafíos
- Evaluación de los marcadores sanguíneos de daño miocárdico
Referencias:
- TIF. TIF. https://thalassaemia.org.cy/
- Salas P. GENÉTICA MOLECULAR APLICADA al DIAGNÓSTICO de ENFERMEDADES HEREDITARIAS TALASEMIAS Caso Clínico: Paciente Con Anemia Microcítica.; 2013. https://www.seqc.es/download/tema/5/2955/3022644/1181332/cms/tema-3-talasemias.pdf/
- Talasemia Y Otras Hemoglobinopatías Informe de La Secretaría PREVALENCIA de LAS HEMOGLOBINOPATÍAS. https://apps.who.int/gb/archive/pdf_files/EB118/B118_5-sp.pdf